CLONACIÓN DEL GEN DE LA INSULINA
INTRODUCCIÓN:
El objetivo de este protocolo será clonar nuestro gen de la insulina, para lo cual debemos abordar una serie de cuestiones, ya que habrá varias opciones para llevar a cabo este experimento, aunque con distintas ventajas e inconvenientes que expondremos en cada decisión que tengamos que tomar:
- ADN o ARN:
Se emplea ADN, ya que el ARN es más inestable y además presenta un inconveniente: deberíamos tomar muestras de células pancreáticas (células β de los islotes de Langerhans) para obtener el ARNm, lo cual complicaría el procedimiento experimental.
- Sangre o saliva:
Como opción, la sangre es más fiable, por lo que acabaremos utilizando la misma para la realización de este proyecto. Esto se debe a que los kits para sangre son más variados y tienen más calidad. La saliva, sin embargo, va a proporcionar menos calidad, además de que los kits son menos variados y el precio total del proceso es más elevado.
El ADN que se va a clonar será el que contienen los leucocitos de la sangre.
Para el protocolo de obtención de la muestra, necesitaremos un dispositivo llamado MagnaRack, el cual compraremos a partir del siguiente enlace:
https://www.ebay.com/c/67898596
1. Obtención de la muestra.
El protocolo elegido de Thermo Fisher tiene un coste de 194 euros por 50 muestras, lo que nos supondría un precio de 3,88 euros para nuestra clonación.
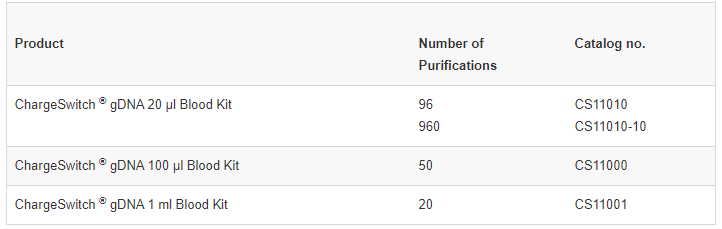
Para realizar la extracción se ha usado unos de los kits de Thermo Fisher, concretamente: ChargeSwitch® gDNA 20 µl Blood Kit, que es el especificado en el protocolo, con número de catálogo CS11010. De momento, este se encuentra descatalogado, por lo que Termo Fisher nos recomienda adquirir el Kit de suero de ADNg de ChargeSwitch™, 0,2-1 ml (CS11040) que contiene los mismos compuestos que el Kit anterior, pero que deberemos diluir diez veces (0,2 ml equivale a 200 µl).
1. El tampón de lisis ChargeSwitch debe diluirse previamente ya que viene más concentrado de lo necesario. El tampón de lisis ChargeSwitch de 20 µl está descontinuado del catálogo, por lo que compraremos el tampón de lisis ChargeSwitch de 0.2-1 ml, como se indicó anteriormente.
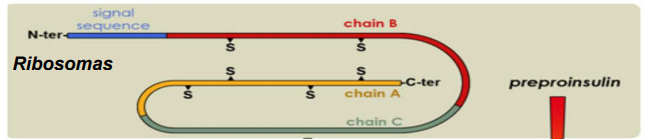
Se emplea ADN, ya que el ARN es más inestable y además presenta un inconveniente: deberíamos tomar muestras de células pancreáticas (células β de los islotes de Langerhans) para obtener el ARNm, lo cual complicaría el procedimiento experimental.
- Sangre o saliva:
Como opción, la sangre es más fiable, por lo que acabaremos utilizando la misma para la realización de este proyecto. Esto se debe a que los kits para sangre son más variados y tienen más calidad. La saliva, sin embargo, va a proporcionar menos calidad, además de que los kits son menos variados y el precio total del proceso es más elevado.
El ADN que se va a clonar será el que contienen los leucocitos de la sangre.
Para el protocolo de obtención de la muestra, necesitaremos un dispositivo llamado MagnaRack, el cual compraremos a partir del siguiente enlace:
https://www.ebay.com/c/67898596
1. Obtención de la muestra.
El protocolo elegido de Thermo Fisher tiene un coste de 194 euros por 50 muestras, lo que nos supondría un precio de 3,88 euros para nuestra clonación.
Para realizar la extracción se ha usado unos de los kits de Thermo Fisher, concretamente: ChargeSwitch® gDNA 20 µl Blood Kit, que es el especificado en el protocolo, con número de catálogo CS11010. De momento, este se encuentra descatalogado, por lo que Termo Fisher nos recomienda adquirir el Kit de suero de ADNg de ChargeSwitch™, 0,2-1 ml (CS11040) que contiene los mismos compuestos que el Kit anterior, pero que deberemos diluir diez veces (0,2 ml equivale a 200 µl).
1. El tampón de lisis ChargeSwitch debe diluirse previamente ya que viene más concentrado de lo necesario. El tampón de lisis ChargeSwitch de 20 µl está descontinuado del catálogo, por lo que compraremos el tampón de lisis ChargeSwitch de 0.2-1 ml, como se indicó anteriormente.
https://diagnosticoencasa.com/producto/dispositivo-de-puncion-toma-de-muestras-sangre-en-diabeticos-100-lancetas/ este viene con pinchador
Esta sección proporciona pautas e instrucciones para aislar DNA genómico de muestras de 10 a 20 µl de sangre humana. Tenga en cuenta que el protocolo está optimizado para una purificación eficaz del DNA de estos volúmenes de muestra.
- Material de partida:
· Antes de empezar:
2. Prepare una mezcla de lisis: para cada muestra, mezcle 0,5 ml de tampón de lisis ChargeSwitch® (L12) y 5 µl de proteinasa K para preparar la mezcla de lisis. Si está aislando ADN de varias muestras, puede aumentar el volumen de reactivos utilizados y preparar una mezcla de lisis maestra. Agite en vórtex el tubo que contiene las perlas magnéticas ChargeSwitch® para resuspender completamente y distribuir uniformemente las perlas en el tampón de almacenamiento.
3. Prepare una mezcla de purificación: para cada muestra, mezcla 20 µl de perlas magnéticas ChargeSwitch (completamente resuspendidas) y 100 µl de tampón de purificación ChargeSwitch (N5) para preparar la mezcla de purificación. Si está aislando DNA de varias muestras, puede aumentar el volumen de reactivos utilizados y preparar una mezcla de purificación maestra.
· Preparación del lisado:
Siga el siguiente procedimiento para preparar un lisado a partir de la muestra de sangre de 10-20 µl.
1. Transfiera la muestra de sangre de 10-20 µl a un tubo de microcentrífuga estéril (o una placa de pocillos profundos de 96 x 2 ml).
2. Añada 0.5 ml de lisis mix a la muestra y pipetee hacia ariiba y hacia abajo suavemente 5 veces para mezclar. Importante: utilice una punta de pipeta de 1 ml ajustada a 450 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar formar burbujas.
3. Incube la muestra a temperatura ambiente durante 10 minutos o hasta que la muestra esté clara y sin grumos visibles.
4. Continúe con la unión de DNA.
· Unión de DNA:
Siga el siguiente procedimiento para unir el DNA a las perlas magnéticas ChargeSwitch.
1. Pipetee suavemente hacia arriba y hacia abajo la mezcla de purificación que contiene las perlas magnéticas Charge Switch para resuspender completamente las perlas.
2. Agregue 120 µl de ChargeSwitch Purification Mix a la muestra dirigida y pipetee hacia arriba y hacia abajo 5 veces para mezclar. Importante: usar una punta de pipeta de 1 ml ajustada a 550 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar que se formen burbujas.
3. Incube a temperatura ambiente durante 1 minuto para permitir que el DNA se una a las perlas magnéticas ChargeSwitch.
4. Coloque la muestra en el MagnaRack (o en el separador magnético de 96 pocillos si utiliza una placa de 96 pocillos profundos) durante 1 minutos o hasta que las perlas formen un sedimento apretado.
5. Sin retirar la muestra del MagnaRack, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no alterar el gránulo de perlas inclinando la pipeta de modo que la punta apunte en dirección opuesta al gránulo.

6. Retire la muestra que contiene las perlas magnéticas granuladas del MagnaRack. No debe haber sobrenadante en el tubo.
7. Añada 500 µl de tampón de lisis ChargeSwitch (L 12, sin proteinasa K) al tubo y pipetee hacia arriba y hacia abajo suavemente 3 veces para mezclar. Utilice una pipeta de 1 ml ajustada a 450 µl.
8. Añada 50 µl de tampón de purificación ChargeSwitch (N5) y pipetee hacia arriba y hacia abajo suabmente 3 veces para mezclar. Utilice una pipeta de 1 ml ajustada a 500 µl.
9. Incubar a temperatura ambiente durante 1 minuto.
10.Coloque la muestra e el MagnaRack (o en el separador magnético de 96 pocillos si crresponde) durante 1 minuto o hasta que las perlas hayan formado un sedimento apretado.
11. Sin retirar la muestra del MagnaRack, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no alterar el gránulo de perlas inclinando la pipeta de modo que la punta apunte en dirección opuesta al gránulo.
12. Proceda inmediatamente a Lavado de DNA, a continuación.
· Lavado de DNA:
1. Retire la muestra que contiene las perlas magnéticas del MagnaRack (paso 11 arriba), No debe haber sobrenadante en el tubo.
2. Añada 500 µl de tampón de lavado ChargeSwitch (W12) a la muestra y pipetee suavamente hacia arriba y hacia abajo dos veces para resuspender las perlas magnéticas. Importante: utilice una punta de pipeta de 1 ml ajustada a 900 µl para mezclar la muestra. Asegúrese de que la punta esté sumergida y pipetee hacia arriba y hacia abajo suavemente para evitar que se formen burbujas.
3. Coloque la muestra en el MagnaRack durante 1 minuto o hasta que las perlas hayan formado un gránulo apretado.
4. Sin retirar la muestra del MagnaRack, retire con cuidado el sobrenadante y deséchelo. Tenga cuidado de no alterar el gránulo de perlas inclinando la pipeta de modo que la punta apunte en dirección opuesta al gránulo.
5. Proceda a eluir el DNA.
· Elución de ADN:
1. Retire la muestra que contiene las perlas magnéticas granuladas del MagnaRack (Paso 4, arriba). No debe haber sobrenadante en el tubo.
2. Añada 100 µl de tampón de elución ChargeSwitch (E5) (o tampón TE, pH 8,5) a la muestra y pipetee hacia arriba y hacia abajo suavemente 10 veces para resuspender las perlas magnéticas. Importante: no utilice agua para la elución. El ADN no eluirá debido a la escasa capacidad amortiguadora del agua.
3. Incubar a temperatura ambiente durante 1 minuto.
4. Coloque la muestra en el MagnaRack durante 3 minutos o hasta que las perlas hayan formado un gránulo apretado.
5. Sin retirar el tubo del MagnaRack, retire con cuidado el sobrenadante que contiene el ADN a un tubo de microcentrífuga estéril (o una placa de microtitulación con fondo en U de 96 x 300 µl). Tenga cuidado de no alterar el gránulo de perlas inclinando la pipeta de modo que la punta apunte en dirección opuesta al gránulo.
6. Deseche las perlas magnéticas usadas. No reutilice las cuentas.
· Almacenamiento de ADN:
Almacene el ADN purificado a -20 ° C o utilícelo inmediatamente para el análisis posterior. Evite congelar y descongelar repetidamente el ADN.
· Cuantificación del rendimiento de ADN:
Para cuantificación de dsDNA Quant-iT PicoGreen (nº de catálogo P7589). Este kit permite cuantificar el rendimiento de su ADN, recomendamos utilizar el kit de contiene los reactivos necesarios para permitir una detección basada en reactivo de cuantificación Quant-iT PicoGreen dsDNA fluorescencia sensible y precisa de tan solo 25 pg / ml de dsDNA.
Para más información sobre el protocolo, clique aquí
Para información audiovisual sobre el protocolo, clique aquí
2. Diseño de los cebadores:
https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?ORGANISM=9606&INPUT_SEQUENCE=AH002844.2&LINK_LOC=nuccore (Sitio web del NCBI para el diseño de cebadores).
Para diseñar los cebadores necesarios para amplificar el gen de la insulina, primero debemos encontrar el gen de la insulina y su secuencia, para lo que utilizaremos la base de datos nucleotídica del NCBI: buscaremos a partir de los términos “insulin” y “human”, donde encontraremos la secuencia siguiente:

Con código de acceso AH002844.2.


Este se forma a raíz de la unión de los dos exones marcados y la eliminación del intrón interior. Concretamente, se encuentran entre las bases 2424-2610, y 3397-3542. Por lo tanto, vamos a clonar la secuencia completa del CDS (1119 nucleótidos de longitud).
El siguiente paso será diseñar los cebadores para esta secuencia elegida, por lo que nos dirigimos a la herramienta de diseño de cebadores del NCBI (enlace al principio del punto) en la que señalaremos nuestro gen (con su código de acceso señalado anteriormente).

(Vemos el código de acceso del gen y el rango de los cebadores, elegidos con un margen de 400 nucleótidos entre el sitio de inicio y fin del CDS).
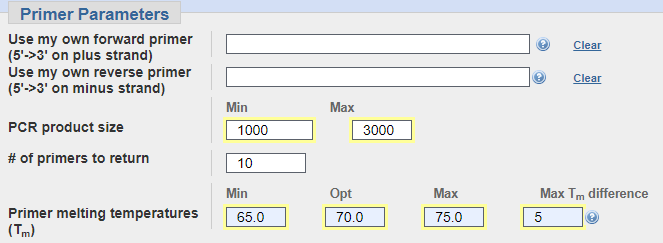
Vemos las temperaturas óptimas y los tamaños de producto de la PCR.
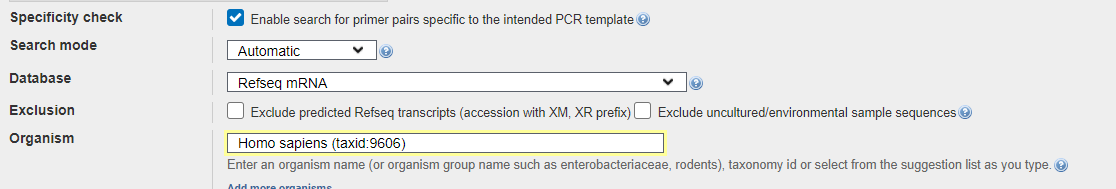
Evitamos que coincida con otra secuencia en humanos (que sea específico)
Obtenemos los resultados:
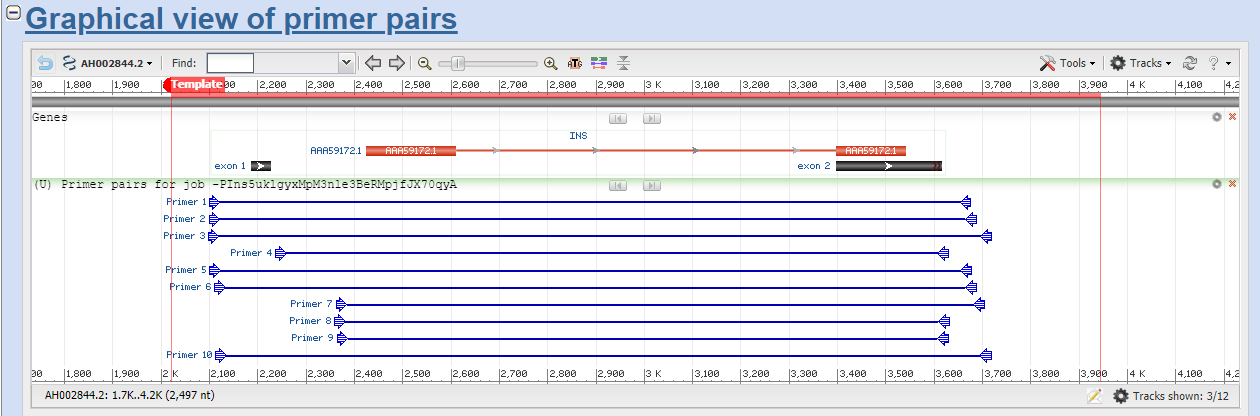
Vemos que el 9 se adapta mejor a la longitud del CDS por lo que escogemos este:
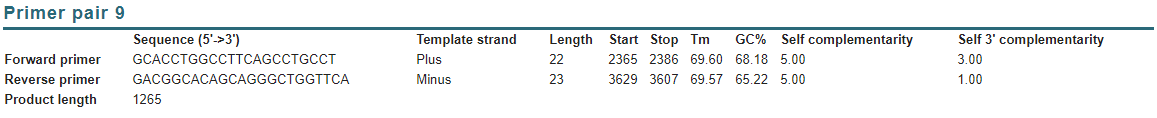
El siguiente paso será calcular la TM para los cebadores:
Calculadora TM:
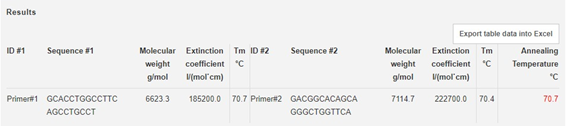
https://www.thermofisher.com/order/custom-standard-oligo
3. Obtención del DNA mediante PCR:
https://currlabcsueb.weebly.com/pcr-polymerase-chain-reaction.html (Protocolo PCR)
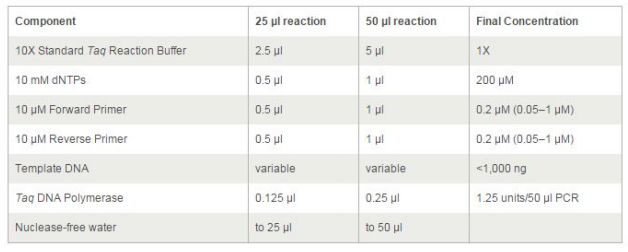
En esta tabla se explica la concentración de reactivos que hay que añadir a unos tubos para
realizar la reacción de la PCR. En el caso de los cebadores (primers)
se indica un rango de concentraciones permisibles que oscilan entre los 0,05 a 1µM.
La cantidad óptima que debería usarse puede calcularse mediante la optimización de la
reacción. Atendiendo a la tabla, también puede hacerse una reacción de PCR en un volumen
final de 25 o de 50 µL, aunque normalmente se realizan en un volumen de 25µL.
En el apartado “Component”
nos referimos a las soluciones “madre” más concentradas y recomendada de los diferentes
componentes de la mezcla para que añadiendo el volumen indicado en la segunda o tercera
columna, se obtenga la concentración final indicada en la cuarta columna.
Por tanto, calcularemos a continuación cómo preparar una disolución madre de 10 micromolar
en cebadores si, la empresa que nos los suministra, nos proporciona un tubo con
25 nanomoles en total de dicho cebador y el tubo es capaz de albergar como máximo 1,5 mL.

En la tabla nos indican la conveniencia de preparar la solución madre de cada uno de los
cebadores a una concentración 10 micromolar.
Disolvente con volumen máximo de 1,5ml.
10 micromolar de disolución madre significa que hay 10 micromoles en un litro, por lo que hay 10^4 nanomoles en un total de 10^3 mL. Al convertir los micromoles en nanomoles, ya podemos utilizar la información que nos han dado de los 25 nanomoles.
Si pudiésemos añadir 2,5 mL de tampón TE (tampón tris-EDTA) a cada uno de esos tubos que nos envían con los 25 nanomoles de cebador, ya conseguimos la solución madre 10 micromolar deseada.
Como la capacidad máxima del tubo del cebador es de 1,5ml, y no podemos añadir los 2,5 mL:
En vez de 2,5 mL de agua (=2500 µl), añadimos 0,25 mL = 250µl de agua. Por tanto, echamos 10 veces menos de lo que tenemos que añadir finalmente, por lo que lo que tendremos en el tubo original es una concentración de cebadores 10X, lo que significa que la concentración que obtendremos estará 10 veces más concentrada. Para comprobarlo, realizamos otra regla de 3:
Si en 250 microL tenemos 25 nmoles, en 10^6 microL (1L), tenemos que tener 100000 nM o, lo que es igual, 100 microM (se confirma que tenemos 10X, ya que 10 x 10 = 100 microM).
Se cierra el tubo y se agita.
Se centrifuga el tubo para que toda la disolución vaya al fondo y no se quede en el tapón.
Como hemos preparado la disolución 10X, a la hora de trabajar con ella deberemos tener en cuenta que está 10 veces concentrada o diluirla 10 veces. Por ejemplo, lo haremos añadiendo 10 microlitros de disolución madre a 90 microL de tampón (de 100 microM pasa a ser 10 microM). Por lo que:
Con esta disolución, obtendremos 100 microL. Si vamos a hacer unos pocos tubos dePCR, también podemos diluir la solución madre añadiendo 1 microL de solución 100 microM de cebadores + 9 microL de tampón.
Esto se puede distribuir en un total de 11 tubos, como se ha mencionado anteriormente (dilución 10+90) por separado, de formaque obtenemos 11 tubos independientes. Si se contamina alguno, tendremos otros 10.
Otra cuestión que debemos realizarnos es:
¿Qué concentración de solución madre de cebadores (la de la columna de Components) debo preparar para que, al añadir 0,5 microL de dicha solución madre, se obtenga en un tubo para PCR con 25 microL una concentración final de cebadores 0,2 microM, tal y como indica la tabla?
Lo primero que vamos a calcular es la cantidad de cebadores que tiene que haber en esos 25 µL de volumen final del tubo de PCR para que haya una concentración final 0,2 µM de cebadores. Esta cantidad es lo que vamos a introducir en cada tubo y de cada cebador para hacer la reacción de PCR.

Por tanto, en un tubo de PCR con 25 µL, hace falta añadir 5 pmoles de cada uno de los cebadores para obtener una concentración final 0,2 µM de cada uno de cada cebador se usa de forma individual (Todo el proceso se repite dos veces, uno para cada cebador). los cebadores.
Según los cálculos realizados, debemos añadir 5 µmoles de cada cebador a los 25 µL para una reacción de PCR, y deben añadirse en un volumen de 0,5 µL. Esto será fácil ya que anteriormente hemos preparado hasta 11 tubos de cada cebador con una concentración 10 µM y un volumen de 100 µL cada uno. Por lo tanto, tomando 0,5 µL de uno de estos tubos, ya estaríamos tomando los 5 pmoles requeridos (0,5 µL * 10 µmol / 10^6 µL = 5 * 10^-6 µmol = 5pmoles).
Estas cantidades podemos calcularlas con el Nanodrop, un dispositivo que mide en segundos muestras muy pequeñas de ADN, ARN o proteínas; las cuales no son medibles de forma exacta con otros utensilios de laboratorio.
También lo utilizaremos para cuantificar la cantidad de ADN molde que añadimos a la muestra (con 1ng es suficiente).
Utilizaremos http://www.endmemo.com/bio/tm.php para calcular ™ una vez tengamos la concentración.
Por último, nos preguntaremos:
¿Cuántas reacciones de PCR con los volúmenes y concentraciones indicadas en la Tabla (25 µL y 0,2 µM de cada uno de los cebadores) podemos llegar a realizar si solicitamos a la empresa fabricante de cebadores que nos suministre 25 nmoles (por defecto) de cada uno de los cebadores?
Para calcular la rentabilidad del proceso vamos a ver cuantas reacciones y por qué precio nos saldrían si tomamos 25 µL de mezcla (Concentración final de reacción es de 0,2 µM).
En el apartado anterior se calculó que, en un tubo de 25 µL, 0,2 µM en cebadores, era necesario suministrar 5 pmoles de cada uno de los cebadores:





Vídeo explicativo de cómo montar e interpretar una PCR

http://www.endmemo.com/bio/tm.php
[Sal]=0
[Mg2+]=1.5mM (según el protocolo)
™=96.6ºC
4. Purificación del ADN obtenido con la PCR:
Para ello haremos una electroforesis, que en nuestro caso será mediante un gel de agarosa. Aquí vemos un vídeo en el que se explica cómo se hace el gel de agarosa para la electroforesis. Además de esto observamos otro vídeo donde podemos ver cómo se realiza la electroforesis tras la preparación del gel de agarosa.
Sin embargo, debemos establecer ciertos parámetros antes de empezar a hacer la electroforesis. En primer lugar, debemos establecer unos marcadores moleculares, de manera que sepamos identificar el ADN de la insulina cuando lo terminemos de separar. Estos marcadores moleculares se deben ver respecto al tamaño de las muestras, de forma que sabiendo que el gen de la insulina tiene una longitud de 1431 nucleótidos la escala que utilizaremos será una que comprenda esa longitud de bases, como por ejemplo es esta.
Protocolo a seguir para hacer correr una electroforesis en gel de agarosa:
Equipo:
- Bandeja de fundición
- Bien peines
- Fuente de voltaje
-Caja de gel
- Fuente de luz ultravioleta
- Microonda
Reactivos:
- TAE
- Agarosa
- Bromuro de etidio (concentración de stock de 10 mg / mL)
Procedimiento
Verter un gel de agarosa estándar al 1%:
1. Mide 1 g de agarosa.
2. Mezcle el polvo de agarosa con 100 ml de TAE 1x en un matraz apto para microondas.
3. Cocine en el microondas durante 1-3 minutos hasta que la agarosa se disuelva por completo (pero no hierva demasiado la solución, ya que parte del tampón se evaporará y, por o tanto, alterará el porcentaje final de agarosa en el gel. Muchas personas prefieren calentar en el microondas en pulsos, haciendo girar el matraz ocasionalmente a medida que la solución se calienta).
4. Dejar la solución de agarosa reposar a aproximadamente 50 ° C (aproximadamente cuando pueda mantener cómodamente la mano en el matraz), aproximadamente 5 minutos.
5. (Opcional)
Agregue bromuro de etidio (EtBr) a una concentración final de aproximadamente
0.2-0.5 μg / mL (generalmente alrededor de 2-3 μl de solución madre de
laboratorio por 100 mL de gel). EtBr se une al ADN y le permite visualizar el
ADN bajo luz ultravioleta (UV).
Agregue bromuro de etidio (EtBr) a una concentración final de aproximadamente
0.2-0.5 μg / mL (generalmente alrededor de 2-3 μl de solución madre de
laboratorio por 100 mL de gel). EtBr se une al ADN y le permite visualizar el
ADN bajo luz ultravioleta (UV).
6. Vierta la agarosa en una bandeja de gel con el peine de pozos en su lugar.
7. Coloque el gel recién vertido a 4 ° C durante 10-15 minutos O déjelo reposar a
temperatura ambiente durante 20-30 minutos, hasta que se haya solidificado por
completo.
Carga de muestras y procesamiento de un gel de agarosa:
1. Agregue tampón de carga a cada una de sus muestras de ADN.
Nota: El tampón de carga tiene dos propósitos: 1) proporciona un tinte visible que ayuda con la carga del gel y le permite medir qué tan lejos ha migrado el ADN; 2) contiene un alto porcentaje de glicerol que aumenta la densidad de su muestra de ADN haciendo que se asiente en el fondo del gel, en lugar de difundirse en el tampón.
2. Una vez solidificado, coloque el gel de agarosa en la caja de gel (unidad de electroforesis).
3. Llene la caja de gel con 1xTAE (o TBE) hasta que el gel esté cubierto.
4. Cargue con cuidado una escalera de peso molecular en el primer carril del gel.
Nota: Cuando cargue la muestra en el pocillo, mantenga una presión positiva sobre la muestra para evitar que entren burbujas o tampón en la punta. Coloque la parte superior de la punta de la pipeta en el tampón justo encima del pocillo. Muy lenta y constantemente, empuje la muestra hacia afuera y observe como la muestra llena el pocillo. Una vez descargada toda la muestra, empuje la pipeta hasta el segundo tope y levante con cuidado la pipeta para sacarla del tampón.
5. Cargue con cuidado sus muestras en los pocillos adicionales del gel.
6. Ejecute el gel a 80-150 V hasta que la línea de tinte esté aproximadamente al 75-80% del recorrido del gel. Un tiempo de ejecución típico es de aproximadamente 1-1,5 horas, dependiendo de la concentración y el voltaje del gel.
Nota: el negro es negativo, el rojo es positivo. El ADN está cargado negativamente y correrá hacia el electrodo positivo. Siempre corre a rojo.
7. Apague la energía, desconecte los electrodos de la fuente de energía y luego retire con cuidado el gel de la caja de gel.
8. (Opcional) Si no agregó EtBr al gel y al tampón, coloque el gel en un recipiente lleno con 100 mL de tampón de ejecución TAE y 5 μL de EtBr, colóquelo en un balancín durante 20-30 minutos, reemplace la solución de EtBr con agua y destiñe durante 5 min.
9. Usando cualquier dispositivo que tenga luz ultravioleta, visualice sus fragmentos de ADN. Los fragmentos de ADN generalmente se denominan "bandas" debido a su apariencia en el gel.
Podemos hacer una valoración de la calidad del resultado obtenido al realizar la electroforesis en gel de agarosa. En este enlace viene explicado al detalle (para más información véase el siguiente vídeo).
Una vez tengamos nuestro ADN unido a la agarosa, el siguiente paso será separarlo de este.
1. Una vez que haya ejecutado su gel, muévalo a una caja UV abierta (asegúrese de usar protección UV adecuada, ¡especialmente para sus ojos!), Retírelo de cualquier bandeja de gel ya que el plástico bloqueará gran parte de los rayos UV y con un paño limpio y estéril. hoja de afeitar, corte el fragmento de ADN deseado del gel.
5. SECUENCIACIÓN DNA:
Los cebadores no se añaden a la vez en la secuenciación, ya que las bases se mezclan y no se distinguen las correspondencias entre las diferentes hebras.
Usaremos, además, un marcador de peso molecular.
La secuenciación se utilizará para observar en la secuencia de nuestro gen las posibles mutaciones que pueda haber en el individuo analizado. Esta técnica, actualmente, se realiza a gran velocidad, pudiéndose requerir a una empresa de secuenciación como la Universidad de Córdoba, en nuestro caso.
Dicha empresa, requiere de diversas cantidades de ADN diluidas en agua para la reacción de secuenciación, las cuales deberemos adjuntar junto con la solicitud. Las cantidades serán pequeñas, por lo que las mediremos con el Nanodrop anteriormente mencionado:

El precio de la secuenciación será de 4,75 euros, tal y como indica la tabla de abajo en esta organización; pero debemos de hacer dos secuenciaciones de cadena simple, ya que una comenzará en la secuencia (sentido forward) y otra, también con el mismo sentido, continuará desde la mitad de la cadena hasta el final (ya que el producto tiene aproximadamente 1200 pb y la secuenciación solo asegura calidad hasta aproximadamente 700 pb), por esto diseñamos un segundo cebador forward hacia la mitad de la secuencia:
Como el primer forward inicial comienza en el pb 2365, este segundo primer debería comenzar en torno al pb 2950-3000:

Tomamos solo el primer Forward que comienza en 2985 y terminaría en 3006. Como el primer cebador debería perder la calidad en el pb 3065 (2365+700), las dos secuencias secuenciadas solapan bien y podríamos secuenciar el CDS de la insulina (en dos partes). (DEBEMOS COMPRARLO IGUAL QUE ANTES)
El precio total de secuenciación, por tanto, es de 9,5 euros.

La empresa nos enviará dos archivos: la secuencia del gen en formato FASTA y un cromatograma en formato .abi, el cual es la medida experimental, con los resultados obtenidos. Dicho cromatograma, el cual incluirá la secuencia de nucleótidos del gen de la insulina clonado, podrá ser analizado por distintos programas, como Chromas, del cual veremos su utilidad y manejo en las entradas posteriores.
Referencias:
1.- https://www.ncbi.nlm.nih.gov/genome/gdv/browser/gene/?id=627
Comentarios
Publicar un comentario